
p_0 <- 0.5 # initial allele frequency ("50% marbles" from lecture #1)
N <- 500 # number of chromosomes in a population
T <- 2000 # number of generations to simulate
# a vector for storing frequencies over time
p_trajectory <- p_0
# in each generation:
for (gen_i in 2:T) {
# get frequency in the previous generation
p_prev <- p_trajectory[gen_i - 1]
# calculate new frequency ("sampling marbles from a jar")
p_trajectory[gen_i] <- rbinom(1, N, p_prev) / N
}
plot(p_trajectory)Simulations in population genetics
August 2023
Many problems in population genetics cannot be solved by a mathematician, no matter how gifted. [It] is already clear that computer methods are very powerful. This is good. It […] permits people with limited mathematical knowledge to work on important problems […].
Making sense of inferred statistics

Making sense of inferred statistics

Fitting model parameters (i.e. ABC)

Ground truth for method development

From Fernando’s lecture on Monday…

\(N\) = 500, \(p_0 = 0.5\)
Code
plot(p_trajectory, type = "l", ylim = c(0, 1),
xlab = "generations", ylab = "allele frequency")
abline(h = p_0, lty = 2, col = "red")
\(N\) = 500, \(p_0 = 0.5\) (20 replicates)
Code
reps <- replicate(20, simulate(N = 500, p_0 = 0.5, T = 2000))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)
\(N\) = 1000, \(p_0 = 0.5\) (20 replicates)
Code
reps <- replicate(20, simulate(N = 1000, p_0 = 0.5, T = 2000))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)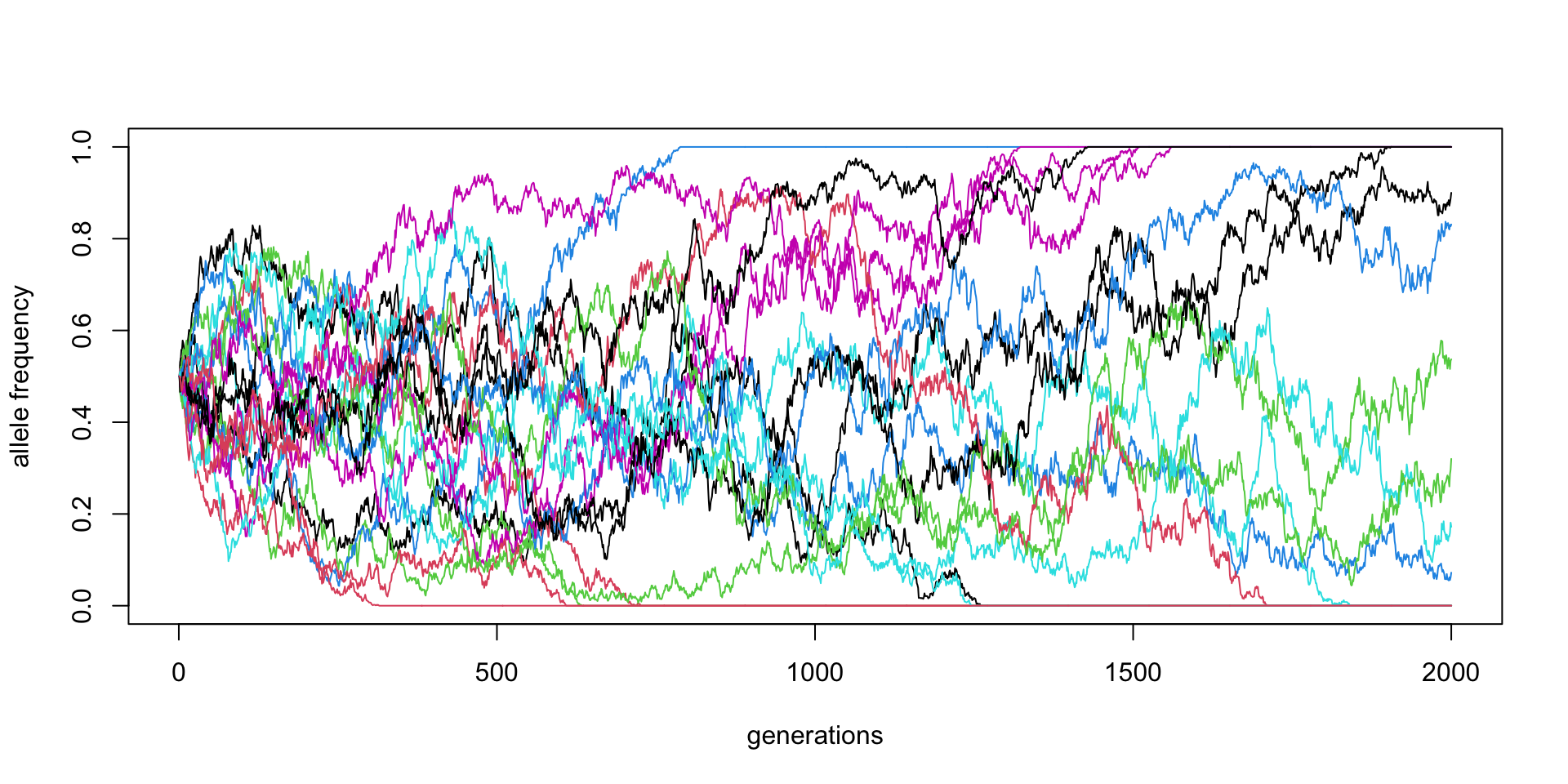
\(N\) = 5000, \(p_0 = 0.5\) (20 replicates)
Code
reps <- replicate(20, simulate(N = 5000, p_0 = 0.5, T = 2000))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)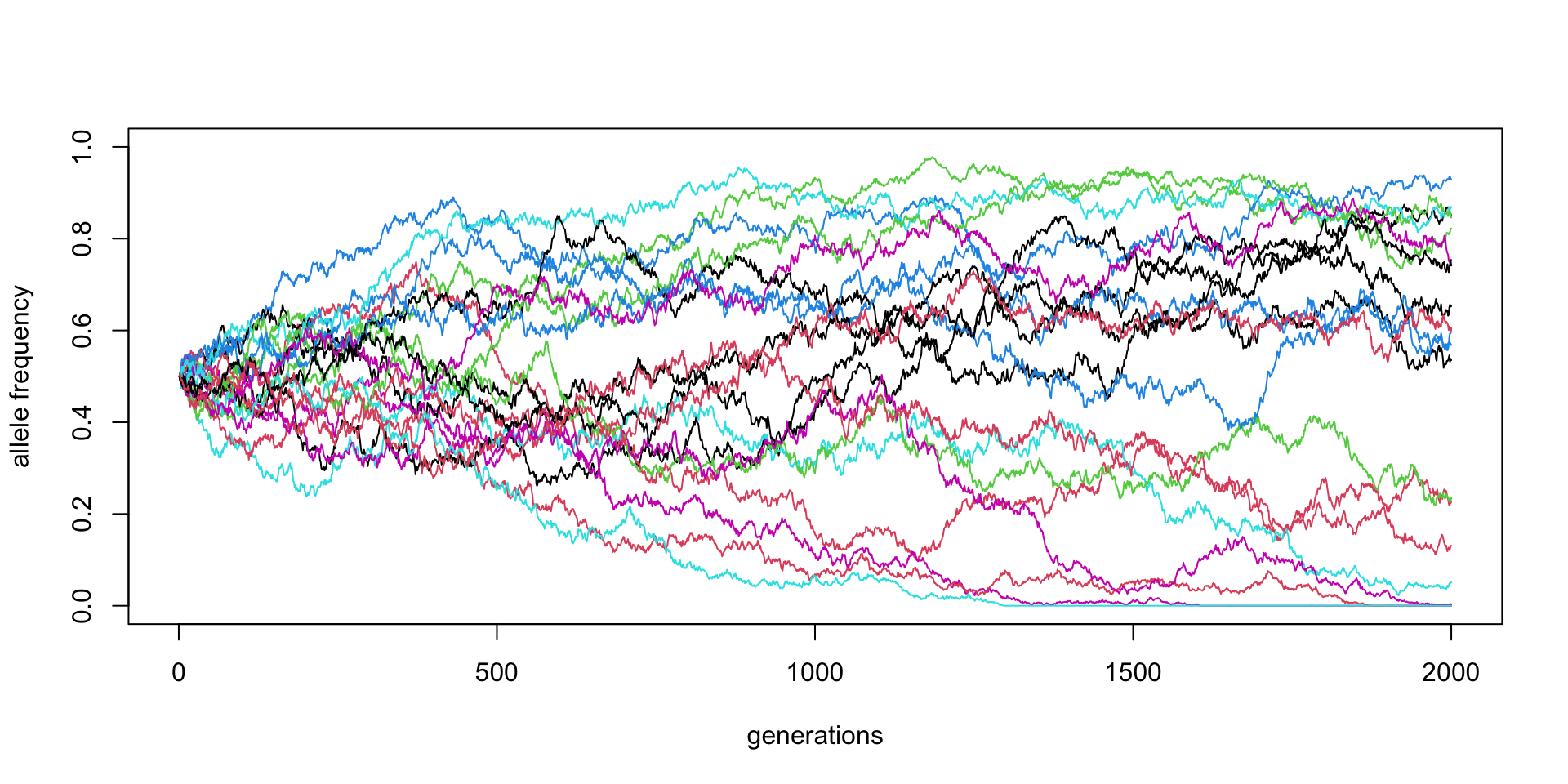
\(N\) = 10000, \(p_0 = 0.5\) (20 replicates)
Code
reps <- replicate(20, simulate(N = 10000, p_0 = 0.5, T = 2000))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)
\(N\) = 10000, \(p_0 = 0.5\) (100 replicates)
Code
reps <- replicate(100, simulate(N = 10000, p_0 = 0.5, T = 30000))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)
\(N\) = 10000, \(p_0 = 0.5\) (100 replicates)
Code
factors <- MASS::fractions(c(3, 2, 1, 1/2, 1/5, 1/10))
matplot(reps, ylim = c(0, 1), xlab = "generations", ylab = "allele frequency", type = "l", lty = 1)
abline(v = 10000 * factors, lwd = 5)
Expected allele frequency distribution
Code
library(ggplot2)
library(dplyr)
library(parallel)
if (!file.exists("diffusion.rds")) {
p_0 <- 0.5
N <- 10000
factors <- MASS::fractions(c(3, 2, 1, 1/2, 1/5, 1/10))
final_frequencies <- parallel::mclapply(
seq_along(factors), function(i) {
f <- factors[i]
t <- as.integer(N * f)
# get complete trajectories as a matrix (each column a single trajectory)
reps <- replicate(10000, simulate(N = N, p_0 = p_0, T = t))
# only keep the last slice of the matrix with the final frequencies
data.frame(
t = sprintf("t = %s * N", f),
freq = reps[t, ]
)
},
mc.cores = parallel::detectCores()
) %>% do.call(rbind, .)
final_frequencies$t <- forcats::fct_rev(forcats::fct_relevel(final_frequencies$t, sprintf("t = %s * N", factors)))
saveRDS(final_frequencies, "diffusion.rds")
} else {
final_frequencies <- readRDS("diffusion.rds")
}
final_frequencies %>% .[.$freq > 0 & .$freq < 1, ] %>%
ggplot() +
geom_histogram(aes(freq, y = after_stat(density), fill = t), position = "identity", bins = 100, alpha = 0.75) +
labs(x = "allele frequency") +
coord_cartesian(ylim = c(0, 3)) +
facet_grid(t ~ .) +
guides(fill = guide_legend(sprintf("time since\nthe start\n[assuming\nN = %s]", N))) +
theme_minimal() +
theme(strip.text.y = element_blank(),
axis.title.x = element_text(size = 15),
axis.title.y = element_text(size = 15),
axis.text.x = element_text(size = 15),
axis.text.y = element_text(size = 15),
legend.title = element_text(size = 15),
legend.text = element_text(size = 15))

What is SLiM?
- A forward-time simulator
- It’s fully programmable!
- Massive library of functions for:
- demographic events
- various mating systems
- selection, quantitative traits, …
- > 700 pages long manual!

What is msprime?
- A Python module for writing coalescent simulations
- Extremely fast (genome-scale, population-scale data)
- You must know Python fairly well to build complex models

www.slendr.net
![]()
Why a new package?
Spatial simulations!

Model visualization
plot_model(model, proportions = TRUE)
Go to the lecure & exercises page
Link: github.com/bodkan/ku-popgen2023
Open the relevant red-highlighted links:

Exercise #1: write this model in slendr

Start a model1.R script in RStudio with this “template”:
library(slendr); init_env()
<... population definitions ...>
<... gene flow definition ...>
model <- compile_model(
populations = list(...),
gene_flow = <...>,
generation_time = 30
)
plot_model(model) # verify visuallyWhat is tree sequence?

- a record of full genetic ancestry of a set of samples
- an encoding of DNA sequence carried by those samples
- an efficient analysis framework
What we usually have

What we usually want
An understanding of our samples’ evolutionary history:
This is exactly what a tree sequence is!
The magic of tree sequences
They allow computing of popgen statistics without genotypes!
There is a “duality” between mutations and branch lengths.
What if we need mutations though?
Coalescent and mutation processes can be decoupled!
What if we need mutations though?
Coalescent and mutation processes can be decoupled!

With slendr, we can add mutations after the simulation using ts_mutate().
Let’s take the model from earlier…

slendr’s R interface to tskit statistics

Allele-frequecy spectrum, diversity \(\pi\), \(F_{ST}\), Tajima’s D, etc.
Find help at slendr.net/reference or in R under ?ts_fst etc.
Example: allele frequency spectrum
Sample 5 individuals:
names <- ts_names(ts)[1:5]
names[1] "pop_1" "pop_2" "pop_3" "pop_4" "pop_5"Compute the AFS:
afs <- ts_afs(ts, list(names))
afs[-1] [1] 4177 2025 1278 969 768 672 552 491 408 340
plot(afs[-1], type = "b",
xlab = "allele count bin",
ylab = "frequency")
Note: We drop the first element (afs[-1]) technical reasons related to tskit. You don’t have to worry about that here, but you can read this for more detail.
Exercise #3: more statistics! (a)
Use msprime() to simulate a 50Mb tree sequence ts from your introgression model in model1.R (if that takes more than two minutes, try just 10Mb).

(Remember to add mutations with ts_mutate().)
Exercise #4a: ancient samples
Let’s return to your introgression model:

Exercise #4b: \(f_4\)-ratio statistic
Use \(f_4\)-ratio statistic to replicate the following figure:
